Type I interferon signaling pathway enhances immune-checkpoint inhibition in KRAS mutant lung tumors
Fernández-García F, Fernández-Rodríguez A, Fustero-Torre C, Piñeiro-Yáñez E, Wang H, Lechuga CG, Callejas S, Álvarez R, López-García A, Esteban-Burgos L, Salmón M, San Román M, Guerra C, Ambrogio C, Drosten M, Santamaría D, Al-Shahrour F, Dopazo A, Barbacid M and Musteanu M.
Lung cancer is the leading cause of cancer mortality worldwide. KRAS oncogenes are responsible for at least a quarter of lung adenocarcinomas, the main subtype of lung cancer. After four decades of intense research, selective inhibitors of KRAS oncoproteins are finally reaching the clinic. Yet, their effect on overall survival is limited due to the rapid appearance of drug resistance, a likely consequence of the high intratumoral heterogeneity characteristic of these tumors. In this study, we have attempted to identify those functional alterations that result from KRAS oncoprotein expression during the earliest stages of tumor development. Such functional changes are likely to be maintained during the entire process of tumor progression regardless of additional co-occurring mutations. Single-cell RNA sequencing analysis of murine alveolar type 2 cells expressing a resident Kras oncogene revealed impairment of the type I interferon pathway, a feature maintained throughout tumor progression. This alteration was also present in advanced murine and human tumors harboring additional mutations in the p53 or LKB1 tumor suppressors. Restoration of type I interferon (IFN) signaling by IFN-β or constitutive active stimulator of interferon genes (STING) expression had a profound influence on the tumor microenvironment, switching them from immunologically "cold" to immunologically "hot" tumors. Therefore, enhancement of the type I IFN pathway predisposes KRAS mutant lung tumors to immunotherapy treatments, regardless of co-occurring mutations in p53 or LKB1.
Unlocking the Genetic Secrets of Pancreatic Cancer: KRAS Allelic Imbalances in Tumor Evolution
Liaki V, Rosas-Perez B, Guerra C.
Pancreatic Ductal Adenocarcinoma (PDAC) belongs to the types of cancer with the highest lethality. It is also remarkably chemoresistant to the few available cytotoxic therapeutic options. PDAC is characterized by limited mutational heterogeneity of the known driver genes, KRAS, CDKN2A, TP53, and SMAD4, observed in both early-stage and advanced tumors. In this review, we summarize the two proposed models of genetic evolution of pancreatic cancer. The gradual or stepwise accumulated mutations model has been widely studied. On the contrary, less evidence exists on the more recent simultaneous model, according to which rapid tumor evolution is driven by the concurrent accumulation of genetic alterations. In both models, oncogenic KRAS mutations are the main initiating event. Here, we analyze the emerging topic of KRAS allelic imbalances and how it arises during tumor evolution, as it is often detected in advanced and metastatic PDAC. We also summarize recent evidence on how it affects tumor biology, metastasis, and response to therapy. To this extent, we highlight the necessity to include studies of KRAS allelic frequencies in the design of future therapeutic strategies against pancreatic cancer.
Navigating the complexities of pancreatic ductal adenocarcinoma: A review on therapeutic models and RAS inhibitors
Barrambana S, Zamorano-Domínguez E, Liaki V, Guerra C.
Pancreatic cancer is one of the most lethal types of cancer, known for a poor prognosis. Currently, the standard of care for unresectable tumors consists of combinations of cytotoxic chemotherapy. Thus far, targeted therapies against specific oncogenic pathways have not been approved for clinical use. Most cases of pancreatic cancer are sporadic/non-hereditary Pancreatic Ductal Adenocarcinomas (PDACs) and are caused by activating mutations in the KRAS oncogene. For the past four decades, KRAS was considered "undruggable". However, numerous multi-selective and mutant-specific RAS inhibitors are now under active development. In this review, we present experimental models of PDAC that facilitate studies of response to therapy and drug resistance. We also discuss recent evidence on targeted therapeutic strategies under preclinical and clinical evaluation, with emphasis on the KRAS signaling.
Kras oncogene ablation prevents resistance in advanced lung adenocarcinomas
Salmón M, Álvarez-Díaz R, Fustero-Torre C, Brehey O, Lechuga CG, Sanclemente M, Fernández-García F, López-García A, Martín-Guijarro MC, Rodríguez-Perales S, Bousquet-Mur E, Morales-Cacho L, Mulero F, Al-Shahrour F, Martínez L, Domínguez O, Caleiras E, Ortega S, Guerra C, Musteanu M, Drosten M and Barbacid M.
KRASG12C inhibitors have revolutionized the clinical management of patients with KRASG12C-mutant lung adenocarcinoma. However, patient exposure to these inhibitors leads to the rapid onset of resistance. In this study, we have used genetically engineered mice to compare the therapeutic efficacy and the emergence of tumor resistance between genetic ablation of mutant Kras expression and pharmacological inhibition of oncogenic KRAS activity. Whereas Kras ablation induces massive tumor regression and prevents the appearance of resistant cells in vivo, treatment of KrasG12C/Trp53-driven lung adenocarcinomas with sotorasib, a selective KRASG12C inhibitor, caused a limited antitumor response similar to that observed in the clinic, including the rapid onset of resistance. Unlike in human tumors, we did not observe mutations in components of the RAS-signaling pathways. Instead, sotorasib-resistant tumors displayed amplification of the mutant Kras allele and activation of xenobiotic metabolism pathways, suggesting that reduction of the on-target activity of KRASG12C inhibitors is the main mechanism responsible for the onset of resistance. In sum, our results suggest that resistance to KRAS inhibitors could be prevented by achieving a more robust inhibition of KRAS signaling mimicking the results obtained upon Kras ablation.
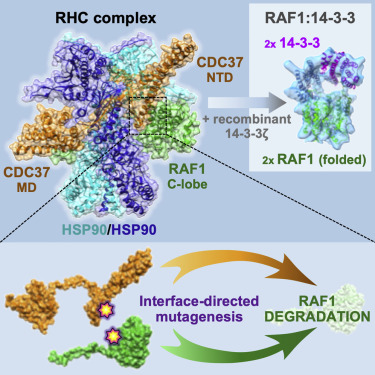
Structure of the RAF1-HSP90-CDC37 complex reveals the basis of RAF1 regulation
García-Alonso S, Mesa P, de la Puente Ovejero L, Aizpurua G, Lechuga CG, Zarzuela E, Santiveri CM, Sanclemente M, Muñoz J, Musteanu M, Campos-Olivas R, Martínez-Torrecuadrada J, Barbacid M, and Montoya G.
Abstract: RAF kinases are RAS-activated enzymes that initiate signaling through the MAPK cascade to control cellular proliferation, differentiation, and survival. Here, we describe the structure of the full-length RAF1 protein in complex with HSP90 and CDC37 obtained by cryoelectron microscopy. The reconstruction reveals a RAF1 kinase with an unfolded N-lobe separated from its C-lobe. The hydrophobic core of the N-lobe is trapped in the HSP90 dimer, while CDC37 wraps around the chaperone and interacts with the N- and C-lobes of the kinase. The structure indicates how CDC37 can discriminate between the different members of the RAF family. Our structural analysis also reveals that the folded RAF1 assembles with 14-3-3 dimers, suggesting that after folding RAF1 follows a similar activation as B-RAF. Finally, disruption of the interaction between CDC37 and the DFG segment of RAF1 unveils potential vulnerabilities in attempting the pharmacological degradation of RAF1 for therapeutic purposes.
Targeting the MAPK Pathway in KRAS-Driven Tumors
Drosten M and Barbacid M
Abstract: KRAS mutations occur in a quarter of all of human cancers, yet no selective drug has been approved to treat these tumors. Despite the recent development of drugs that block KRASG12C, the majority of KRAS oncoproteins remain undruggable. Here, we review recent efforts to validate individual components of the mitogenactivated protein kinase (MAPK) pathway as targets to treat KRAS-mutant cancers by comparing genetic information derived from experimental mouse models of KRAS-driven lung and pancreatic tumors with the outcome of selective MAPK inhibitors in clinical trials. We also review the potential of RAF1 as a key target to block KRAS-mutant cancers.
RAF1 kinase activity is dispensable for KRAS/p53 mutant lung tumor progression
Sanclemente M, Nieto P, García-Alonso S, Fernández-García F, Esteban-Burgos L, Guerra MC, Drosten M, Caleiras E, Martínez-Torrecuadrada J, Santamaría D, Musteanu M and Barbacid M

